Η ΑΡΡΥΘΜΙΟΓΟΝΟΣ ΜΥΟΚΑΡΔΙΟΠΑΘΕΙΑ (ACM) ΚΑΙ Η ΝΟΣΟΣ ΤΗΣ ΝΑΞΟΥ
 ΚΑΙ Η ΝΟΣΟΣ ΤΗΣ ΝΑΞΟΥ")
Ενημερώθηκε στις 4/10/2023
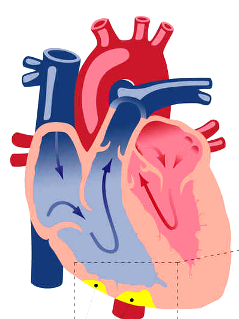
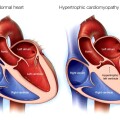
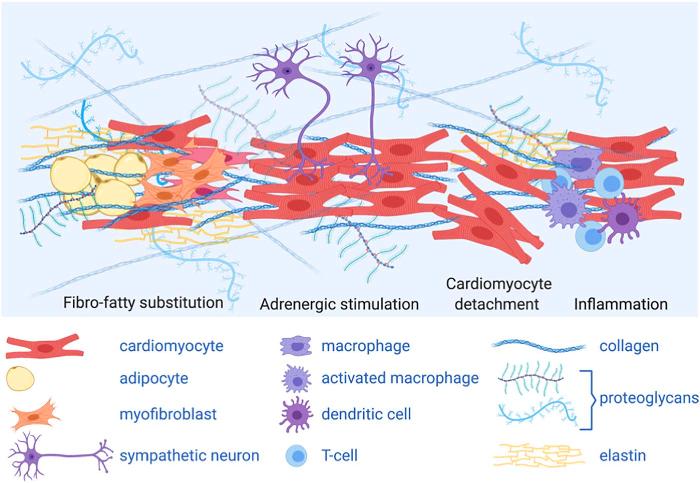
Η αρρυθμιογόνος μυοκαρδιοπάθεια (Arrhythmogenic CardioMyopathy ή ACM) είναι μια πολύπλοκη γονιδιακή – κληρονομική πρωτοπαθής νόσος του καρδιακού μυ όπου τα μυοκαρδιακά κύτταρα προοδευτικά απομακρύνονται το ένα από άλλο και αργότερα πεθαίνουν (απόπτωση), οπότε στη θέση τους δημιουργείται ουλή από ινώδη και ινολιπώδη ιστό.
Το αποτέλεσμα είναι ότι στην αρρυθμιογόνο μυοκαρδιοπάθεια (ACM) προκαλούνται μετά την ηλικία των 10 ετών κακοήθεις κοιλιακές αρρυθμίες που οδηγούν σε συγκοπτικά επεισόδια ή αιφνιδίο θανάτο κυρίως σε ανθρώπους κάτω των 40 ετών, ιδίως σε άθληση, ενώ αργότερα εμφανίζεται ο ινολιπώδης ιστός και δημιουργείται τοπική ή/και σφαιρική δυσλειτουργία της κοιλίας και τελικά καρδιακή ανεπάρκεια (> 40% των ασθενών).
[πρωτοπαθής = δεν οφείλεται σε βλάβη από άλλη αιτία π.χ. από ισχαιμία, βαλβιδοπάθεια, υπέρταση κλπ.]
Η αρρυθμιογόνος μυοκαρδιοπάθεια (ACM) οφείλεται σε γονιδιακή μετάλλαξη, συνηθέστερα σε κάποιο πρόγονο και σπανιότερα σποραδική, που αφορά τη δημιουργία παθολογικής ή μειωμένης κάποιας από τις πρωτεΐνες που συνδέουν δυο γειτονικά κύτταρα του μυοκαρδίου μεταξύ τους και κυρίως των δεσμοσωμάτων και σπάνια αφορά άλλες πρωτεΐνες των παρεμβαλλόμενων δίσκων. (Δες πιο κάτω)
>> Πάντως ακόμη δεν έχει βρεθεί το υπεύθυνο γονίδιο σε ποσοστό περίπου 40% των ασθενών.
(Περίπου στο 5% των περιπτώσεων ACM υπάρχουν περισσότερες από μια παθολογικές συνδετικές πρωτεΐνες.)
Η επίπτωση της ACM στο γενικό πληθυσμό κυμαίνεται από 1/1.000 έως 1/2.000, ανάλογα με την περιοχή. (Για τη νόσο της Νάξου δες πιο κάτω)
Λόγω του ότι τα μυοκαρδιακά κύτταρα απομακρύνονται το ένα από άλλο (από βλάβη σε κάποια από τις πρωτεΐνες που τα συνδέουν), σε πρώτη φάση σταματά η συντονισμένη διάδοση του ηλεκτρικού ρεύματος στο μυοκάρδιο (οπότε δημιουργούνται κοιλιακές αρρυθμίες) και η συντονισμένη καρδιακή σύσπαση (με τοπική δυσλειτουργία*) και σε επόμενη φάση τα μυοκαρδιακά κύτταρα πεθαίνουν (απόπτωση) και στη θέση τους δημιουργείται ινώδης και ινολιπώδης ιστός (ουλή). (Δες πιο κάτω)
Η ACM στο τέλος προκαλεί καρδιακή ανεπάρκεια με μείωση του κλάσματος εξωθήσεως.
(Η *τοπική δυσλειτουργία σε κοιλία εκδηλώνεται με ακινησία, υποκινησία ή παράδοξη κίνηση κυρίως του ελεύθερου τοιχώματος (ή/και του μεσοκοιλιακού διαφράγματος).
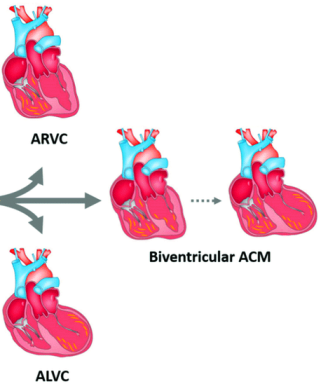
Η αρρυθμιογόνος μυοκαρδιοπάθεια (ACM) εντοπίζεται συνήθως στο μυοκάρδιο και των δυο κοιλιών και αποτελεί εξέλιξη του προηγούμενου όρου της Αρρυθμιογόνου μυοκαρδιοπάθειας της δεξιάς κοιλίας (Arrhythmogenic Right Ventricular Cardiomyopathy ή ARVC). Υπ’ όψιν ότι η ARVC στο 60% της έχει αμφικοιλιακή εντόπιση.
[Παλιότερα η νόσος ονομαζόταν Αρρυθμιογόνος δυσπλασία της δεξιάς κοιλίας (Arrhythmogenic Right Ventricular Dysplasia ή ARVD)
Στις τελευταίες Ευρωπαϊκές οδηγίες του 2023 προτείνεται ένας νέος υπότυπος Μυοκαρδιοπάθειας, η Μη διατεταμένη μυοκαρδιοπάθεια της Αριστερής κοιλίας (Non-Dilated Left Ventricular Cardiomyopathy ή NDLVC).
https://academic.oup.com/eurheartj/article/44/37/3503/7246608
Αυτή περιλαμβάνει τον φαινότυπο της αντικατάστασης μυοκαρδιακού ιστού στην αριστερή κοιλία από ουλή ή/και λίπος, χωρίς όμως να υπάρχει διάταση της.
Αυτός ο φαινότυπος περιλαμβάνει κυρίως την καθ’ υπεροχήν (dominant) Αρρυθμιογόνο μυοκαρδιοπάθεια της αριστερής κοιλίας (ALVC).
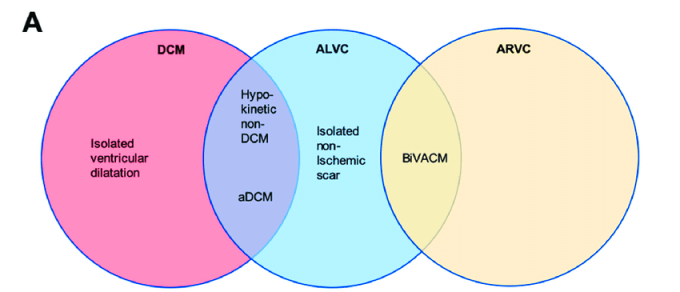
Έτσι διαχωρίζεται (και καταργείται) η αρρυθμιογόνος μυοκαρδιοπάθεια (ACM), στην Αρρυθμιογόνο μυοκαρδιοπάθεια της αριστερής κοιλίας (ALVC) και στην Αρρυθμιογόνο μυοκαρδιοπάθεια της δεξιάς κοιλίας (ARVC Arrhythmogenic Right Ventricular Cardiomyopathy ή ARVC)]
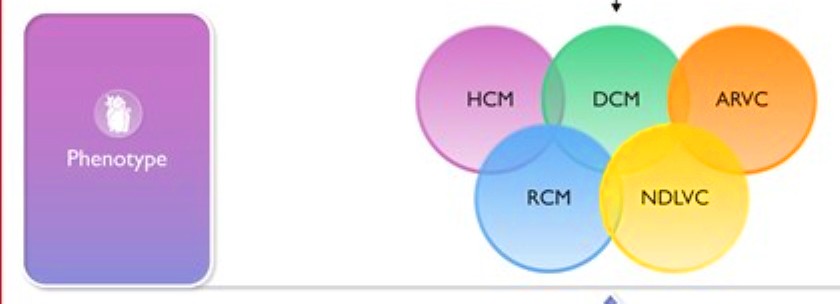
>> Έτσι ο όρος ACM περιλαμβάνει τώρα την καθ’ υπεροχήν (dominant) ARVC, την καθ’ υπεροχήν (dominant) Αρρυθμιογόνο μυοκαρδιοπάθεια της αριστερής κοιλίας (ALVC) και την αμφικοιλιακή ACM (biVentricular ACM).
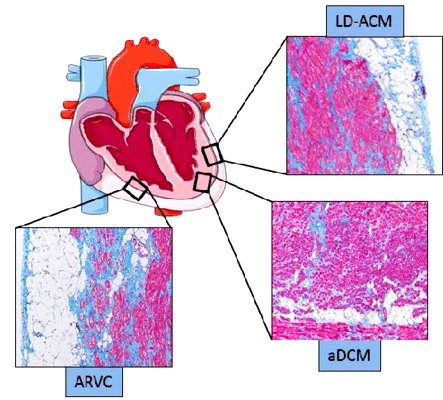
Επιπλέον στην ACM μπορεί να υπάρχει και επικάλυψη με την αρρυθμιογόνο διατατική μυοκαρδιοπάθεια (arrhythmogenic Dilated CardioMyopathy ή aDCM) που είναι υπότυπος της DCM, όπως και με ορισμένες διαυλοπάθειες (π.χ. σε ασθενείς με σύνδρομο Brugada έχουν βρεθεί μεταλλάξεις και στο γονίδιο PKP2 ή με το γονίδιο SCN5A της εισόδου του Νατρίου στο κύτταρο κατά την εκπόλωση του, που σχετίζεται με το σύνδρομο LQTS τύπου 3 και με το σύνδρομο Brugada).
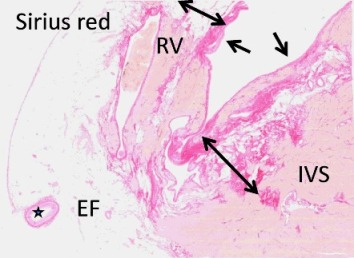
Σε ARVC, αντικατάσταση του μυϊκού ιστού από ινώδη και ινολιπώδη ιστό στο πρόσθιο τοίχωμα της δεξιάς κοιλίας (RV) και στο δεξιό μεσοκοιλιακό διάφραγμα (IVS). (EF = Επικάρδιο λίπος / Ο αστερίσκος δείχνει τη στεφανιαία αρτηρία)
https://onlinelibrary.wiley.com/doi/full/10.1002/joa3.12021
ΣΥΜΠΤΩΜΑΤΑ
Εκδηλώνεται συνήθως μεταξύ 12 και 45 ετών με αίσθημα παλμών, συγκοπή ή και αιφνιδίο θανάτο (ο οποίος μπορεί να αποτελεί την πρώτη εκδήλωση της νόσου).
Σε προχωρημένα στάδια μπορεί να οδηγήσει σε δεξιά καρδιακή ανεπάρκεια και στο τέλος πιθανά και καρδιακή ανεπάρκεια της αριστερής κοιλίας.
Η ΔΙΑΓΝΩΣΗ
Η διάγνωση της ACM είναι ιδιαίτερα δύσκολη.
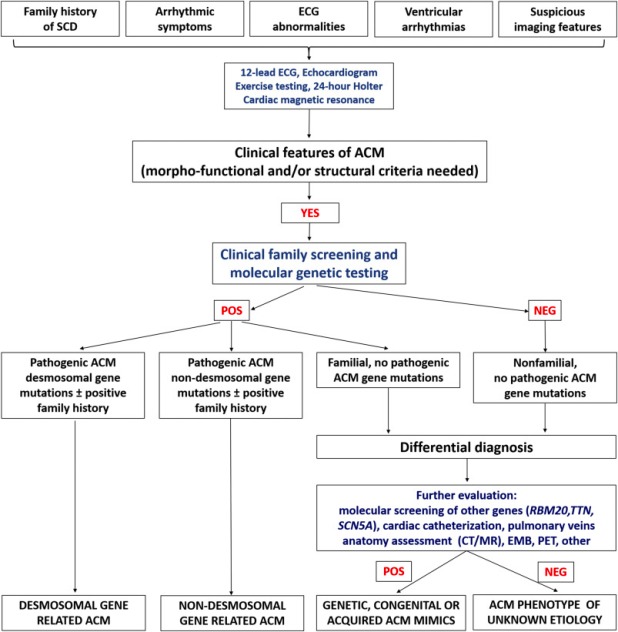
## Θα την υποπτευθεί ο γιατρός σε ηλικίες > 10 ετών με κοιλιακές κυρίως αρρυθμίες, ιδίως αν δημιουργούνται σε σωματική κόπωση (πρώτη εκδήλωση της νόσου) ή με ανεξήγητη δυσλειτουργία της δεξιάς ή/και αριστερής κοιλίας (εκδηλώνεται αργότερα) και ύποπτο ΗΚΓφημα. (δες πιο κάτω)
[Οι κοιλιακές αρρυθμίες μπορεί να είναι έκτακτες κοιλιακές συστολές (πάνω από 500 – 1000/24ωρο) ή κοιλιακή ταχυκαρδία επιμένουσα ή μη, χωρίς να υπάρχει άλλη αιτία που να τις δημιουργεί (αυτές αποτελούν αρχική εκδήλωση της νόσου). Επίσης μπορεί να υπάρχουν κολπικές αρρυθμίες, ή κολποκοιλιακοί αποκλεισμοί.]
## Επίσης θα την υποπτευθεί ο γιατρός αν υπάρχει ιστορικό συγκοπτικού επεισοδίου και τα πιο πάνω ευρήματα.
## Η υποψία θα είναι έντονη, ιδίως αν υπάρχει συγγενής με βεβαιωμένη αρρυθμιογόνο μυοκαρδιοπάθεια (ACM) ή ανεξήγητο αιφνίδιο θάνατο (σε 1ου βαθμού συγγενή) σε ηλικία κάτω των 35 ετών.
Θα ληφθεί ατομικό και οικογενειακό (3 γενεών) ιστορικό και θα διενεργηθεί ΗΚΓμα, υπερηχοκαρδιογράφημα, δοκιμασία κοπώσεως (για έλεγχο δημιουργίας κοιλιακών αρρυθμιών), Holter ΗΚΓφήματος.
Όμως οι σπουδαιότερες εξετάσεις για επιβεβαίωση της νόσου είναι η καρδιακή μαγνητική τομογραφία (CMR) με σκιαγραφικό και τεχνική καθυστερημένης ενίσχυσης (Late Gadolinium Enhancement LGE) όπου ανιχνεύεται ο ινώδης ιστός και τελικά ο γονιδιακός έλεγχος.
** Περισσότερες πληροφορίες για τη διάγνωση υπάρχουν πιο κάτω.
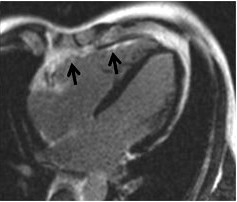
ARVC: Σε καρδιακή μαγνητική τομογραφία υπάρχει διάχυτη LGE στο ελεύθερο τοίχωμα της δεξιάς κοιλίας (Επιμήκης τομή)
https://jcmr-online.biomedcentral.com/articles/10.1186/s12968-014-0050-8/figures/5
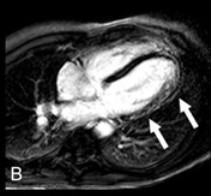
ALVC: Σε καρδιακή μαγνητική τομογραφία υπάρχει διάχυτη LGE στο πλάγιο τοίχωμα και κορυφή της αριστερής κοιλίας (Επιμήκης τομή)
https://www.ahajournals.org/doi/full/10.1161/CIRCEP.111.962779
ΔΙΑΦΟΡΙΚΗ ΔΙΑΓΝΩΣΗ
Σε ήπιες μορφές ARVC χωρίς σαφείς δομικές διαταραχές της δεξιάς κοιλίας, απαιτείται διαφορική διάγνωση από άλλες αρρυθμιογόνες καταστάσεις με δομικά φυσιολογική καρδιά, όπως η κοιλιακή ταχυκαρδία από το χώρο εξόδου της δεξιάς κοιλίας, η κατεχολαμινεργική κοιλιακή ταχυκαρδία και το σύνδρομο Brugada.
Για τη διάγνωση της Αρρυθμιογόνου μυοκαρδιοπάθειας μόνο της αριστερής κοιλίας (ALVC) χρειάζεται να αποκλειστούν άλλες παθήσεις που προσβάλλουν το μυοκάρδιο της αριστερής κοιλίας, όπως η διατατική μυοκαρδιοπάθεια (DCM), η Μυοκαρδίτιδα, η Σαρκοείδωση, η Αμυλοείδωση, η νόσος του Chaga, ορισμένες νευρομυϊκές γονιδιακές νόσοι κλπ.]
ΤΑ ΜΕΤΑΛΛΑΓΜΕΝΑ ΓΟΝΙΔΙΑ
Το γονίδιο που έχει μεταλλαχθεί, συνηθέστερα σε κάποιο πρόγονο, αφορά κυρίως τη δημιουργία κάποιας παθολογικής ή μειωμένης πρωτεΐνης των δεσμοσωμάτων και λιγότερο συχνά από άλλες πρωτεΐνες που δεν σχετίζονται με τα δεσμοσώματα.
Στο 42% περίπου των ασθενών με ACM δεν ανευρίσκεται κάποιο από τα γνωστά μέχρι στιγμής γονίδια.
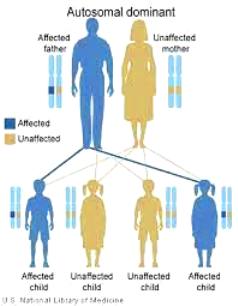
Η κληρονομικότητα της ARVC είναι κυρίως αυτοσωματική επικρατούσα με μειωμένη διεισδυτικότητα (δεν υπάρχει σε όλους τους απογόνους η νόσος*), εκτός της Νόσου της Νάξου (δες πιο κάτω) όπου η μετάλλαξη του γονιδίου JUP κληρονομείται με αυτοσωματικό υπολειπόμενο τρόπο και διεισδυτικότητα >90%.
Η μειωμένη διεισδυτικότητα οφείλεται σε επιγενετικούς παράγοντες και πιθανόν σε άλλα τροποποιητικά γονίδια.
Η αρρυθμιογόνος μυοκαρδιοπάθεια (ACM) μπορεί να οφείλεται σε μετάλλαξη διαφόρων γονιδίων των δεσμοσωμάτων, το δε γονίδιο που βρίσκεται συχνότερα (περίπου στο 65%) μεταλλαγμένο είναι το PKP2 (πλακοφυλλίνη 2) στη δεξιά και αμφικοιλιακή ACM και ακολουθούν το DSG2 (δεσμογλεΐνη 2), το DSP (δεσμοπλακίνη) και πιο σπάνια το DSC2 (δεσμοκολλίνη 2) και το JUP ( πλακοσφαιρίνη) π.χ. στη νόσο της Νάξου.
** Περισσότερες πληροφορίες για τα μεταλλαγμένα γονίδια υπάρχουν πιο κάτω.

Η ΘΕΡΑΠΕΙΑ
Ο στόχος της θεραπείας της ACM είναι η μείωση του κινδύνου του αιφνιδίου θανάτου, η μείωση των αρρυθμιών και η αποτροπή της εξέλιξη της ACM σε καρδιακή ανεπάρκεια.
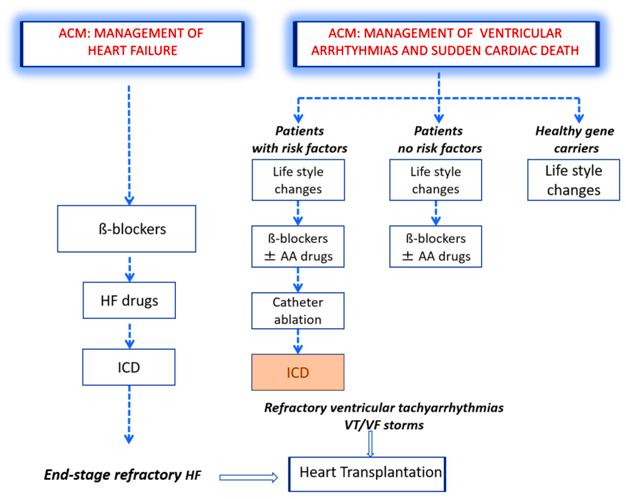
1) Η πρόληψη σε όσους έχουν μόνο μεταλλαγμένο γονίδιο, χωρίς εμφανή νόσο, αφορά μόνο στη μείωση της σωματικής άσκησης.
2) Όσοι έχουν εμφανίσει τη νόσο αντιμετωπίζονται ανάλογα με τον κίνδυνο αιφνιδίου θανάτου που διατρέχουν. Ο κίνδυνος για σοβαρές κοιλιακές αρρυθμίες βασίζεται σε προηγούμενα συμβάντα και συγκεκριμένους παράγοντες κινδύνου.
Η “θεραπεία” περιλαμβάνει:
α) Μείωση της σωματικής άσκησης, β-αναστολείς και πιθανώς αντιφλεγμονώδη φάρμακα.
[>> Συνιστάται μείωση της σωματικής άσκησης ακόμη και αν βρεθεί μόνο μεταλλαγμένο γονίδιο χωρίς εμφανή νόσο = ΟΧΙ ανταγωνιστική ή υψηλής έντασης και διάρκειας άσκηση.
## Η έντονη άσκηση προδιαθέτει σε κοιλιακές αρρυθμίες και επιπλέον γρηγορότερη και βαρύτερη εκδήλωση ACM.
>> Τα αντιφλεγμονώδη πιθανώς μειώνουν τη βλάβη του μυοκαρδίου και τον κίνδυνο αιφνιδίου θανάτου.
(Βρέθηκε ότι καρδιακά μυϊκά κύτταρα λόγω της απομάκρυνσης μεταξύ τους, εκκρίνουν κυτταροκίνες που δημιουργούν φλεγμονή και προσέλκυση κυρίως Τ λεμφοκυττάρων.)
>> Οι β-αναστολείς σε όσους έχουν εκδηλώσει τη νόσο, μειώνουν τον κίνδυνο αρρυθμιών, μειώνουν τη συσπαστικότητα της κοιλίας και πιθανώς μειώνουν τον αιφνίδιο θάνατο.]
β) Αν οι κοιλιακές αρρυθμίες προκαλούν συμπτώματα και ο β-αναστολέας (π.χ. Μετοπρολόλη) δεν επαρκεί, μπορεί να συγχορηγηθεί Αμιοδαρόνη – Angoron (οδηγία ΙΙβ) ή Flecainide – Flecarythm, αν είναι φυσιολογικό το κλάσμα εξωθήσεως (οδηγία ΙΙβ) ή ίσως Σοταλόλη μόνη της (οδηγιά ΙΙβ).
γ) Αblation (κατάλυση της αρρυθμιογόνου εστίας) διενεργείται (μόνο για καλύτερη ποιότητα ζωής) κυρίως αν υπάρχει επαναλαμβανόμενη επιμένουσα ΜΟΝΟΜΟΡΦΗ κοιλιακή ταχυκαρδία (ή ακόμη και μη επιμένουσα) που δεν ανταποκρίνεται στη λήψη β-αναστολέα ή/και Αμιωδαρόνης (οδηγία ΙΙα).
[Η Αblation θα διενεργηθεί με συνδυασμένη επικάρδια και ενδοκάρδια προσέγγιση και με τη βοήθεια ηλεκτροανατομικής χαρτογράφησης.]
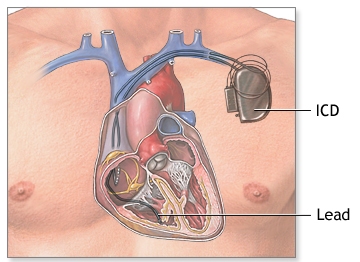
δ) Αυτόματος απινιδωτής (ICD) τοποθετείται σε όσους συμπτωματικούς έχουν ψηλό κίνδυνο αιφνιδίου θανάτου, δηλαδή κυρίως σε όσους είχαν καρδιακή ανακοπή ή είχαν επιμένουσα κοιλιακή ταχυκαρδία ή παρουσίασαν συγκοπτικό αρρυθμιολογικό επεισόδιο ή έχουν μεγάλη μείωση της σύσπασης της δεξιάς ή της αριστερής κοιλίας με κλάσμα εξωθήσεως < από 35%, ιδίως αν είναι άρρενες ηλικίας < 35 ετών.
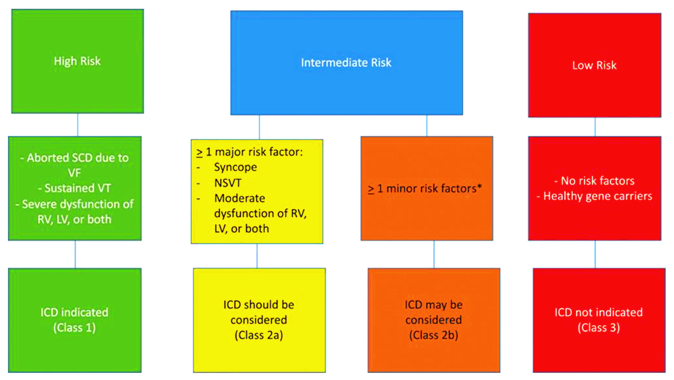
## Για όσους με ARVC έχουν εμφανίσει τη νόσο υπάρχει ένα βοήθημα στο διαδίκτυο που υπολογίζει τον 5ετή πρωτογενή κίνδυνο, για αιφνίδιο θάνατο ή συγκοπτικό επεισόδιο (κοιλιακή ταχυκαρδία, κοιλιακή μαρμαρυγή), στη διεύθυνση: https://arvcrisk.com/
https://academic.oup.com/eurheartj/article/40/23/1850/5419784
[>> Πρόσφατα δημοσιεύτηκε μια μελέτη για την εμφάνιση του αιφνιδίου θανάτου σε ARVC, όπου παραδόξως το μειωμένο κλάσμα εξωθήσεως και η προηγούμενη επιμένουσα κοιλιακή ταχυκαρδία δεν βρέθηκε να αυξάνουν τον κίνδυνο αιφνιδίου θανάτου.
Στη μελέτη βρέθηκαν μόνο 4 κλινικοί παράγοντες για εμφάνιση αιφνιδίου θανάτου σε ARVC: Η μικρή ηλικία, το αντρικό φύλο, η έκταση των αρνητικών Τ στο ΗΚΓφημα και ο αριθμός των εκτάκτων κοιλιακών συστολών.
https://www.ahajournals.org/doi/10.1161/CIRCEP.120.008509
>> Έτσι αρχίζει να έχει κανείς αμφιβολίες για το πότε πραγματικά είναι χρήσιμος ο αυτόματος απινιδωτής (ICD) στην πρωτογενή πρόληψη.]

ε) Για να μειωθεί ο αριθμός των αχρείαστων απινιδώσεων από υπερκοιλιακές ταχυκαρδίες θα υπάρξει περιορισμός σωματικής άσκησης και θα χορηγηθεί ταυτόχρονα και β-αναστολέας (π.χ. Καρβεντιλόλη) με ή χωρίς άλλα αντιαρρυθμικά φάρμακα (π.χ. Αμιοδαρόνη ή Φλεκαινίδη). Για τον ίδιο λόγο μπορεί να χρειαστεί Αblation.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4521905/
ζ) Σε ύπαρξη ανευρύσματος της κοιλίας (ακόμη και χωρίς κολπική μαρμαρυγή) χρειάζεται μακροχρόνια αντιπηκτική θεραπεία, αν βρεθεί θρόμβος στην κοιλία ή αν υπήρξε θρομβοεμβολικό επεισόδιο (αρτηριακό ή φλεβικό).
η) Σε ύπαρξη καρδιακής ανεπάρκειας χορηγείται η ανάλογη θεραπεία, στη δε ARVC χορηγούνται πιθανώς και Νιτρώδη επιπλέον, για μείωση του προφορτίου της δεξιάς κοιλίας.
ΝΟΣΟΣ ΤΗΣ ΝΑΞΟΥ
Μια μορφή της αρρυθμιογόνου μυοκαρδιοπάθειας της δεξιάς κοιλίας εντοπίστηκε στη Νάξο και ονομάστηκε νόσος της Νάξου από τον καρδιολόγο κ. Ν. Πρωτονοτάριο και την Α. Τσατσοπούλου που την πρωτοπαρατήρησαν.
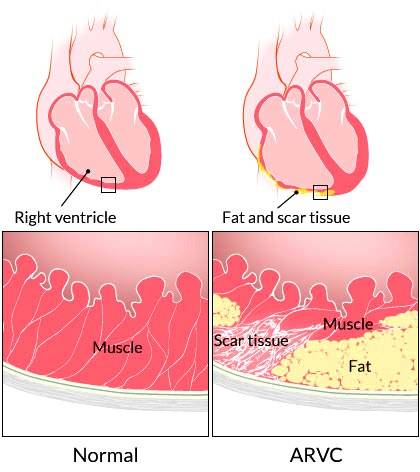
Η νόσος της Νάξου είναι μια μορφή ARVC που πέρα από την αντικατάσταση του μυϊκού με ινολιπώδη ιστό, παρουσιάζει ορισμένα εμφανή χαρακτηριστικά, όπως κερατόδερμα παλαμών και πελμάτων και κατσαρά μαλλιά και επιπλέον μεγαλύτερη διάταση της δεξιάς κοιλίας από τις άλλες μορφές της ARVC.
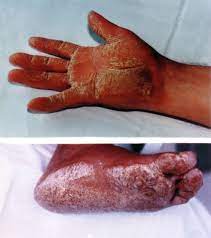
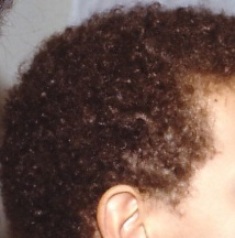
Τα κατσαρά μαλλιά εμφανίζονται με τη γέννηση, το κερατόδερμα των παλαμών και των πελμάτων εμφανίζεται αργότερα, στον 1ο χρόνο της ζωής και η μυοκαρδιοπάθεια γίνεται εμφανής με την ενηλικίωση, με πρώτο σύμπτωμα στους περισσότερους συγκοπτικό επεισόδιο.
Η πρόγνωση της δεν είναι καλή και παρατηρείται αιφνίδιος θάνατος στους νέους σε ποσοστό 2.3% ανά έτος.
Υπ’ όψιν ότι στο 25% των περιπτώσεων η νόσος έχει αμφικοιλιακή εντόπιση.
Η νόσος της Νάξου οφείλεται σε μετάλλαξη του γονιδίου JUP της πρωτεΐνης Πλακοσφαιρίνης (παλιότερα ονομαζόταν γ-Κατενίνη).
Αυτή κληρονομείται με αυτοσωματικό υπολειπόμενο τρόπο (και διεισδυτικότητα πάνω από 90%), δηλαδή αν είναι και οι δύο γονείς ετεροζυγώτες φορείς (έχουν από ένα μεταλλαγμένο γονίδιο JUP), υπάρχει πιθανότητα 25% να γεννηθεί παιδί με τη νόσο, 50% πιθανότητα να γεννηθεί το παιδί σαν φορέας και 25% πιθανότητα το παιδί να μην έχει κανένα παθολογικό γονίδιο JUP.
Οι ετεροζυγώτες φορείς γονείς ίσως παρουσιάζουν μόνο κατσαρά μαλλιά.
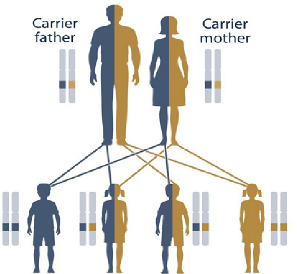
Η συχνότητα των φορέων του μεταλλαγμένου γονιδίου στη Νάξο, είναι για τους ετεροζυγώτες 1 ανά 20 κατοίκους και για τους νοσούντες ομοζυγώτες (και τα δύο γονίδια, ένα από τον πατέρα και ένα από τη μητέρα είναι μεταλλαγμένα) είναι 1 ανά 600 κατοίκους.
Γενικά στις Κυκλάδες η νόσος παρατηρείται σε 1/1000 κατοίκους. Επίσης παρατηρείται στην Τουρκία, στο Ισραήλ κλπ.
[Το σύνδρομο Carvajal είναι παρόμοιο εμφανισιακά και κληρονομικά με τη νόσο της Νάξου, με τη διαφορά ότι το μεταλλαγμένο γονίδιο (DSP) αφορά τη δεσμοπλακίνη και αφορά περισσότερο τη αριστερή κοιλία (ALVC) με μεγάλη διάταση της από την παιδική ηλικία.]
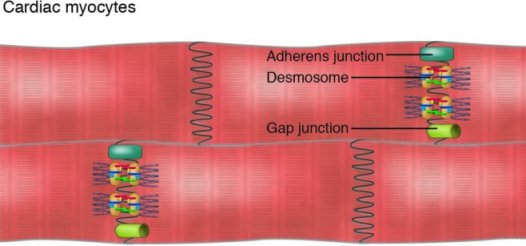
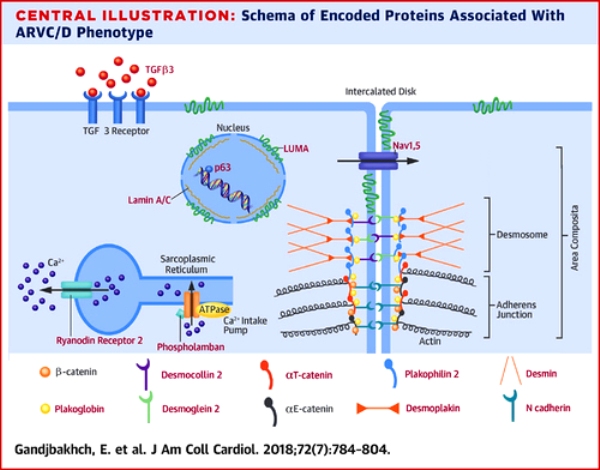
ΟΙ ΠΑΡΕΜΒΑΛΛΟΜΕΝΟΙ ΔΙΣΚΟΙ ΚΑΙ ΤΑ ΔΕΣΜΟΣΩΜΑΤΑ ΑΝΑΜΕΣΑ ΣΤΑ ΜΥΟΚΑΡΔΙΑΚΑ ΚΥΤΤΑΡΑ
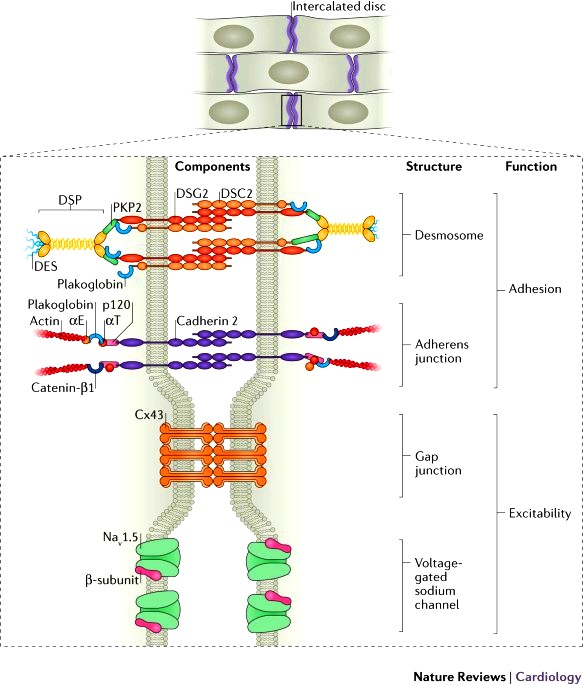
Ο *παρεμβαλλόμενος δίσκος (intercalated disc) συνδέει δυο γειτονικά καρδιακά κύτταρα του μυοκαρδίου και αποτελείται από τα δεσμοσώματα, τους χασμα-συνδέσμους (gap junctions) και τους συνδέσμους πρόσδεσης(adherens junctions). Επιπλέον βρίσκονται σ’ αυτούς δίαυλοι διέλευσης ιόντων.
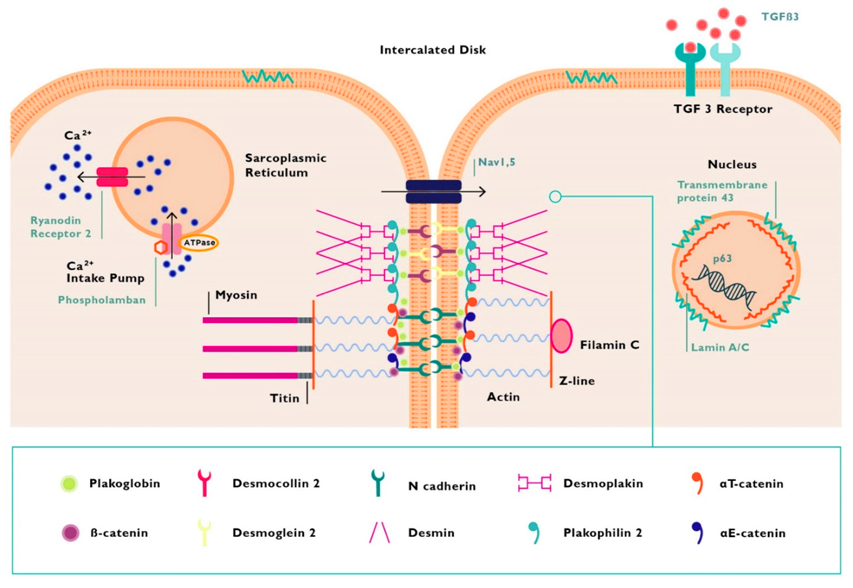
(Τα δεσμοσώματα, οι χασμα-σύνδεσμοι (gap junctions) και οι δίαυλοι διέλευσης ιόντων Νατρίου (Nav 1.5) αποτελούν το συνδεσμόσωμα (connexome) που συνδέει μηχανικά, μεταβολικά και ηλεκτρικά δυο γειτονικά καρδιακά μυϊκά κύτταρα.)
# Τα **δεσμοσώματα είναι σχηματισμοί που συνδέουν δυο γειτονικά κύτταρα μεταξύ τους, μέσω των ενδιάμεσων ινιδίων του κυτταροσκελετού του κάθε κυττάρου, ώστε να διατηρείται η μηχανική ακεραιότητα του ιστού.
Αυτά βρίσκονται στα κύτταρα οργάνων και ιστών που υφίστανται μεγάλη μηχανική καταπόνηση, όπως στην καρδιά, την ουροδόχο κύστη, το δέρμα, τον γαστρεντερικό βλεννογόνο κλπ.
Η κλασική καθ’ υπεροχήν (dominant) ARVC οφείλεται κυρίως σε μεταλλάξεις των γονιδίων των πρωτεϊνών των δεσμοσωμάτων.
# Οι χασμα-σύνδεσμοι ή gap junctions (απαρτίζονται από την πρωτεΐνη κοννεξίνη 43) συνδέουν το κυτταρόπλασμα δυο γειτονικών κυττάρων χημικά – μεταβολικά και ηλεκτρικά.
# Οι σύνδεσμοι πρόσδεσης (adherens junctions) συνδέουν δυο γειτονικά κύτταρα μεταξύ τους μέσω της Ακτίνης τους.
ΠΕΡΙΣΣΟΤΕΡΑ ΓΙΑ ΤΗ ΔΙΑΓΝΩΣΗ ΤΗΣ ACM
Σε όσους δεν βρίσκεται συγγενής με τη νόσο (σποραδική νόσος) και επίσης δεν βρίσκεται και υπεύθυνο γονίδιο που να την προκαλεί, αλλά η υποψία για σποραδική ACM συνεχίζει να υπάρχει, θα γίνει ενδομυοκαρδιακή βιοψία ή οποία αν δείξει τη νόσο τίθεται η διάγνωση (ενώ αν είναι φυσιολογική δεν αποκλείεται η ACM). Επιπλέον η βιοψία θα αποκλείσει άλλες μυϊκές παθήσεις π.χ. τη σαρκοείδωση, τη μυοκαρδίτιδα κλπ
Για τη διάγνωση της δεξιάς, της αριστερής και της αμφικοιλιακής ACM έχουν προταθεί πρόσφατα τα κριτήρια της Πάδουα (Padua) στα οποία δίνεται μεγάλη βαρύτητα στον τελικό φαινότυπο της ανεύρεσης ινώδους ή ινολιπώδους ιστού στο μυοκάρδιο (κυρίως με LGE σε CMR).
Η ΔΙΑΓΝΩΣΗ θα τεθεί σύμφωνα με τα απαραίτητα και ειδικά (μείζονα) κριτήρια της Πάδουα:
>> Είτε με ανεύρεση του μεταλλαγμένου γονιδίου.
Δυστυχώς στο 40-45% των περιπτώσεων της νόσου, δεν ανευρίσκεται γνωστό μεταλλαγμένο γονίδιο, όμως η εύρεση υπεύθυνου γονιδίου με σιγουριά μεγαλύτερη από 90% επιβεβαιώνει την κλινική διάγνωση.
Αν το μεταλλαγμένο γονίδιο βρεθεί, θα διενεργηθεί γονιδιακός έλεγχος και στους συγγενείς, όμως όσον αφορά τα παιδιά χρειάζεται προσεκτική απόφαση, για το καλύτερο συμφέρον τους.
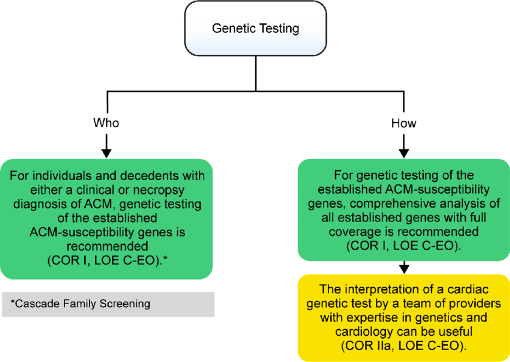
>> Eίτε αν σε ενδομυοκαρδιακή βιοψία (ΕΜΒ) βρεθεί ουλώδης ιστός σε αντικατάσταση μυϊκού ιστού.
>> Είτε με ύπαρξη βεβαιωμένης ACM σε πρώτου βαθμού συγγενή, ΚΑΙ:
> Σε Δεξιά εντόπιση (ARVC):
α) Μπορεί να βρεθεί ινολιπώδης ιστός σε καρδιακή μαγνητική τομογραφία (CMR) με σκιαγραφικό και Late Gadolinium Enhancement (LGE) με γραμμοειδή μορφή σε τουλάχιστον 1 περιοχή του προσθίου τοιχώματος του μυοκαρδίου της δεξιάς κοιλίας (διατοιχωματικά).
β) Σε υπερηχοκαρδιογραφική μελέτη μπορεί να φανεί η ανεύρεση ανευρύσματος, ακινησίας, δυσκινησίας + σφαιρική διάταση της δεξιάς κοιλίας ή μείωση του κλάσματος εξωθήσεως της (<35%).
> Σε Αριστερή μόνο εντόπιση (ALVC):
Μπορεί να βρεθεί ινολιπώδης ιστός σε καρδιακή μαγνητική τομογραφία (CMR) με σκιαγραφικό και Late Gadolinium Enhancement (LGE) με γραμμοειδή μορφή σε τουλάχιστον 1 περιοχή του μυοκαρδίου (υποεπικάρδια και πλαγιοκατώτερα).
[Για τη διάγνωση της ALVC μάλλον χρειάζεται επιπλέον, και η ανεύρεση γονιδίου που την προκαλεί. Επίσης πρέπει να αποκλειστεί η Διατατική μυοκαρδιοπάθεια.]
https://www.sciencedirect.com/science/article/pii/S0167527320332939
Η Ευρωπαϊκή Καρδιολογική Εταιρία το 2010 εξέδωσε οδηγίες για την ARVC. Η διάγνωση θεωρήθηκε σίγουρη, αν υπήρχαν είτε 2 μείζονα, είτε 1 μείζων και 2 ελάσσονα, είτε 4 ελάσσονα κριτήρια, από διαφορετικές κατηγορίες.
ΤΟ ΗΚΓμα
Σε ARVC:
Μπορεί να υπάρχουν αρνητικά Τ κύματα στις V1- V3, χωρίς να υπάρχει πλήρης αποκλεισμός του δεξιού σκέλους (QRS >120 ms), σε ηλικία > 14 ετών (μείζον κριτήριο).
Μπορεί να φαίνεται κύμα Έψιλον στο 13% των περιπτώσεων (έλασσον κριτήριο).
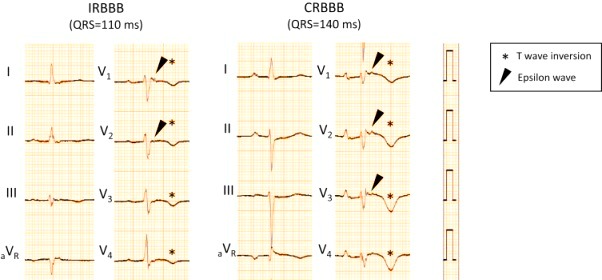
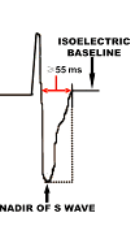
Μπορεί να υπάρχει παράταση του διαστήματος TAD ≥55 ms (από το ναδίρ του S ως το τέλος της εκπόλωσης , δηλαδή την ισοηλεκτρική γραμμή), στις απαγωγές V1- V3 (έλασσον κριτήριο).
Σε ALVC:
Μπορεί να υπάρχουν χαμηλά QRS (<0.5 mV) στις απαγωγές των άκρων, χωρίς όμως να υπάρχει παχυσαρκία ή περικαρδίτιδα ή εμφύσημα (έλασσον κριτήριο).
Μπορεί να υπάρχουν αρνητικά Τ κύματα στις V4- V6, χωρίς να υπάρχει πλήρης αποκλεισμός του αριστερού σκέλους (έλασσον κριτήριο).
ΠΕΡΙΣΣΟΤΕΡΑ ΓΙΑ ΤΑ ΜΕΤΑΛΛΑΓΜΕΝΑ ΓΟΝΙΔΙΑ
Η αρρυθμιογόνος μυοκαρδιοπάθεια (ACM) μπορεί να οφείλεται σε μετάλλαξη διαφόρων γονιδίων των δεσμοσωμάτων, το δε γονίδιο που βρίσκεται συχνότερα (περίπου στο 65%) μεταλλαγμένο είναι το PKP2 (πλακοφυλλίνη 2) στη δεξιά και αμφικοιλιακή ACM και ακολουθούν το DSG2 (δεσμογλεΐνη 2), το DSP (δεσμοπλακίνη) και πιο σπάνια το DSC2 (δεσμοκολλίνη 2) και το JUP ( πλακοσφαιρίνη) π.χ. στη νόσο της Νάξου.
Το μεταλλαγμένο γονίδιο DSP μπορεί να προκαλέσει αριστερή και αμφικοιλιακή ACM (π.χ. το σύνδρομο Carvajal), το μεταλλαγμένο γονίδιο DSG2 μπορεί να προκαλέσει και τις 3 μορφές, ενώ οι άλλες μεταλλαγμένες πρωτεΐνες των δεσμοσωμάτων προκαλούν δεξιά και αμφικοιλιακή ACM.
Οι μεταλλαγμένες ΜΗ δεσμοσωματικές πρωτεΐνες κληρονομούνται με αυτοσωματική επικρατούσα κληρονομικότητα και προκαλούν αμφικοιλιακή ACM και ταυτόχρονα οι περισσότερες αριστερή ACM, π.χ. η lamin A/C (LMNA), η desmin (DES), οι δίαυλοι Νατρίου (SCN5A) (και σύνδρομο Brugada ή μακρού QT ή οικογενή κολπική μαρμαρυγή), η phospholamban (PLN), η filamin C (FLNC) κλπ.
Η μεταλλαγμένη Τιτίνη (TTN) προκαλεί δεξιά και αριστερή ACM, ενώ η μεταλλαγμένη Alpha T-catenin (CTNNA3), ο transforming grow factor beta-3 (TGFβ-3) η N-cadherin (CDH2), και η transmembrane protein 43 (TMEM 43) προκαλούν δεξιά ACM.
Ο μεταλλαγμένος cardiac ryanodine receptor-2 (RYR2) προκαλεί CPVT και διατατική μυοκαρδιοπάθεια.
ΕΝΔΕΙΚΤΙΚΗ ΒΙΒΛΙΟΓΡΑΦΙΑ
https://www.frontiersin.org/articles/10.3389/fphys.2020.568535/full
Arrhythmogenic Cardiomyopathy—Current Treatment … – MDPI
https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.115.017944
https://academic.oup.com/eurheartj/article/40/23/1850/5419784
https://www.intechopen.com/online-first/naxos-disease-current-knowledge-and-future-advances
https://academic.oup.com/eurheartj/article/41/14/1414/5602183
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5946438/
https://www.sciencedirect.com/science/article/pii/S0735109701015686?via%3Dihub
https://www.ahajournals.org/doi/10.1161/CIRCEP.120.008509
https://www.mdpi.com/1422-0067/21/17/6320/htm
https://www.mdpi.com/1422-0067/21/18/6615/htm
https://www.sciencedirect.com/science/article/pii/S0167527320332939
https://www.heartrhythmjournal.com/article/S1547-5271(19)30438-2/fulltext
https://www.jacc.org/doi/10.1016/j.jacc.2018.05.065
https://academic.oup.com/eurheartj/article/41/14/1414/5602183
https://onlinelibrary.wiley.com/doi/10.1111/bjd.19342
https://www.cell.com/current-biology/comments/S0960-9822(11)00477-5
https://www.jacc.org/doi/pdf/10.1016/j.jacc.2014.04.035
https://www.sciencedirect.com/science/article/pii/S0167527320332939
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8268983/